Data on pruritus & fatigue
Jump to:
Data on reduction of pruritus with IQIRVO1
IQIRVO was studied in patients with PBC with moderate-to-severe pruritus, defined as a PBC WI-NRS score ≥4. The change in pruritus from baseline through weeks 24 and 52 were secondary endpoints of the study1
Results with IQIRVO presented below were not statistically significant and, therefore, these results are descriptive only.1
A trend toward improving pruritus was observed with IQIRVO, as measured by WI-NRS (LS mean change from baseline -1.93 vs -1.15 with UDCA alone; 95% CI: -2.0 to 0.4).1
Pruritus impact vs UDCA alone was measured by PBC-40 and 5-D itch scales1,a
PBC-40 itch domain at week 521


The PBC-40 is a patient-derived measure validated for PBC, covering 6 domains: fatigue, pruritus, cognitive, emotional, social, and other symptoms.2
A 0.5-point change per question in the itch domain was a threshold for response, with ≥1.5 change being clinically meaningful.2,3
5-D itch score at week 521


The 5-D itch scale is a patient-reported measure of the degree, duration, direction, disability, and distribution of pruritus.4
aSix patients in the IQIRVO group took IQIRVO alone, while 2 patients in the UDCA group took placebo alone.5
Data on reduction
of fatigue with IQIRVO6
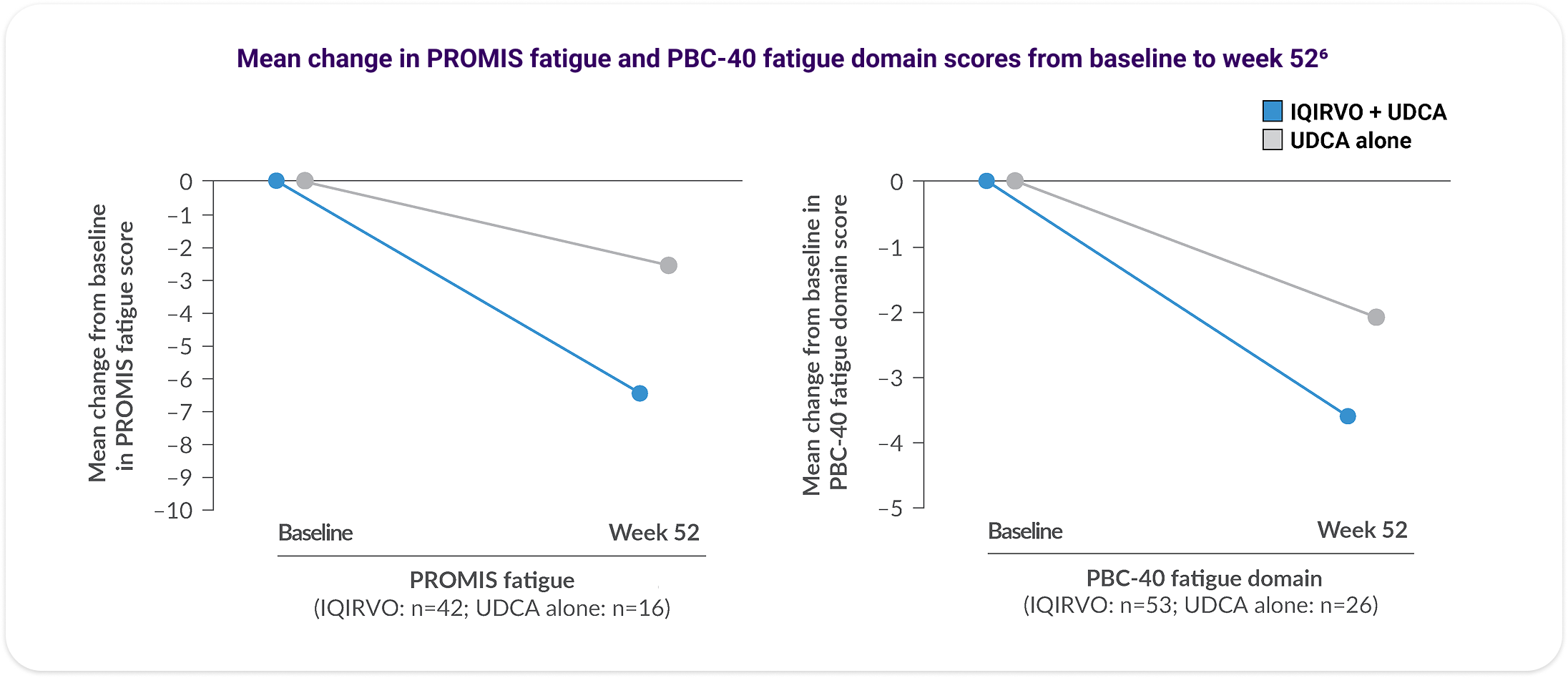
Patients on IQIRVO with moderate-to-severe fatigue reported improvement over time6
An analysis of fatigue via the PRO Measurement Information System (PROMIS) Fatigue Short Form 7a (PROMIS fatigue) and PBC-40 fatigue domain was performed. This was conducted during the ELATIVE® double-blind period and in IQIRVO-treated patients who completed the double-blind period and entered the OLE.6
Decreases observed in PROMIS fatigue and PBC-40 fatigue domain scores in patients with moderate-to-severe fatigue at baseline6
Data are reported for patients with valid fatigue data at baseline and at week 52 and for the subgroup of patients with moderate-to-severe fatigue at baseline (defined as PROMIS fatigue score ≥60 or PBC-40 fatigue domain score ≥29).6 No formal statistical analysis was conducted and results are descriptive only.

Additional PROMIS fatigue and PBC 40 fatigue data from the OLE at weeks 104 and 1306
Changes in fatigue were summarized from baseline to week 104 and week 130 with respect to available MCID, categorical changes, and mean change from baseline in total score.6
The data are from a single arm of the OLE study, including only patients on IQIRVO during ELATIVE. No formal statistical analysis was conducted and results are descriptive only.
Mean change from baseline in total scores of the PROMIS fatigue and PBC-40 fatigue domain6,b




bPatients included are those with non-missing data at baseline and each time point of interest. Moderate-to-severe fatigue was defined as a PROMIS fatigue total score ≥60 or PBC-40 fatigue domain total score ≥29 at baseline. Week 52: n=95, week 104: n=48, week 130: n=26. Dotted lines reflect MCID for each measure.6
CI=confidence interval; LS=least squares; MCID=minimal clinically important difference; OLE=open-label extension; PBC=primary biliary cholangitis; UDCA=ursodeoxycholic acid; WI-NRS=worst itch numeric rating scale.
References: 1. Kowdley KV, Bowlus CL, Levy C, et al; ELATIVE Study Investigators’ Group. Efficacy and safety of elafibranor in primary biliary cholangitis. N Engl J Med. 2024;390(9):795-805, suppl. 2. Jacoby A, Rannard A, Buck D, et al. Development, validation, and evaluation of the PBC-40, a disease specific health related quality of life measure for primary biliary cirrhosis. Gut. 2005;54(11):1622-1629. 3. Jones D, Carbone M, Invernizzi P, et al. Impact of setanaxib on quality of life outcomes in primary biliary cholangitis in a phase 2 randomized controlled trial. Hepatol Commun. 2023;7(3):e0057. 4. Elman S, Hynan LS, Gabriel V, et al. The 5-D itch scale: a new measure of pruritus. Br J Dermatol. 2010;162(3):587-593. 5. IQIRVO [package insert]. Ipsen Biopharmaceuticals, Inc. Cambridge, MA. 6. Data on file. Ipsen Biopharmaceuticals, Inc.